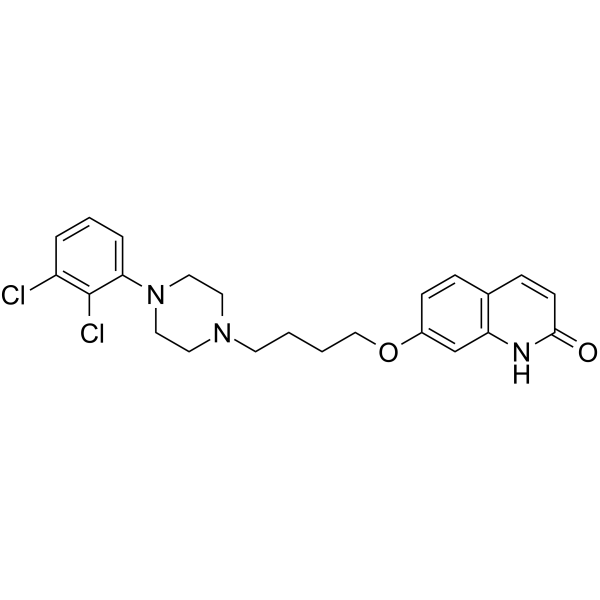
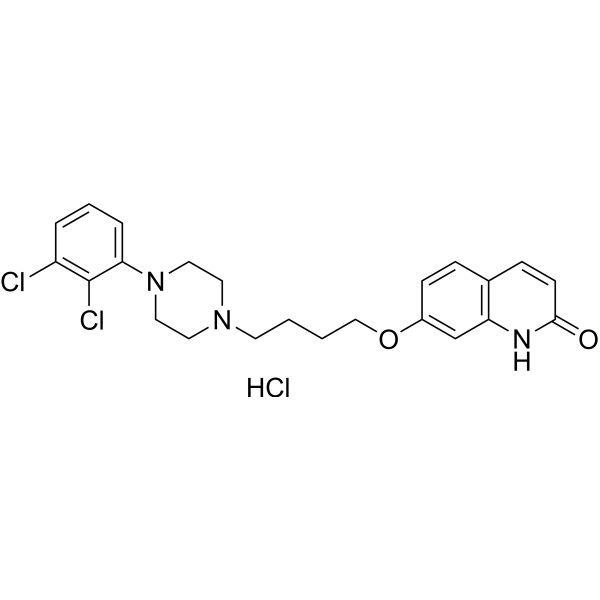
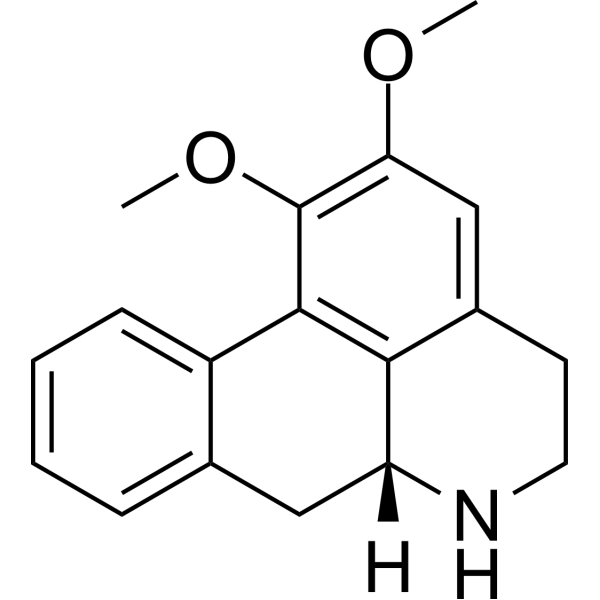
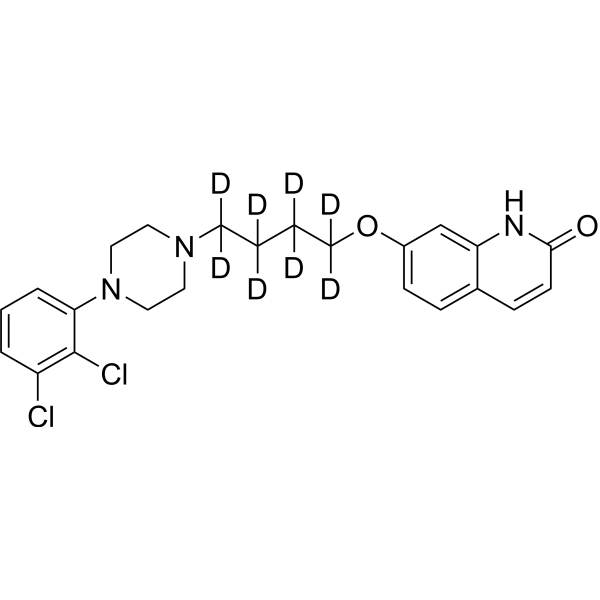
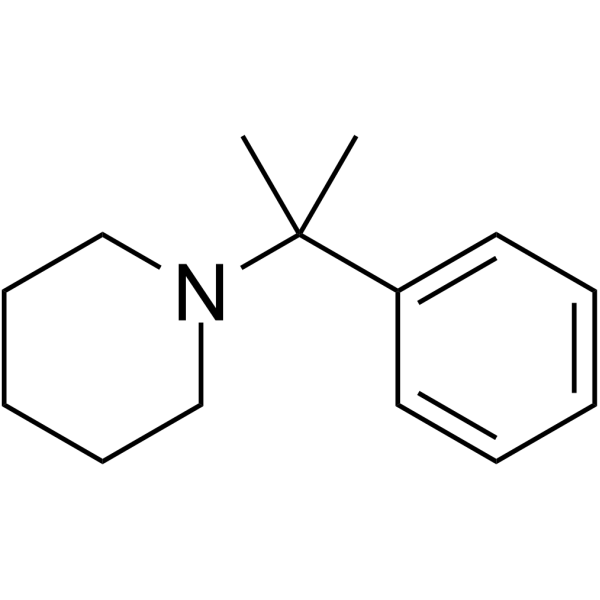
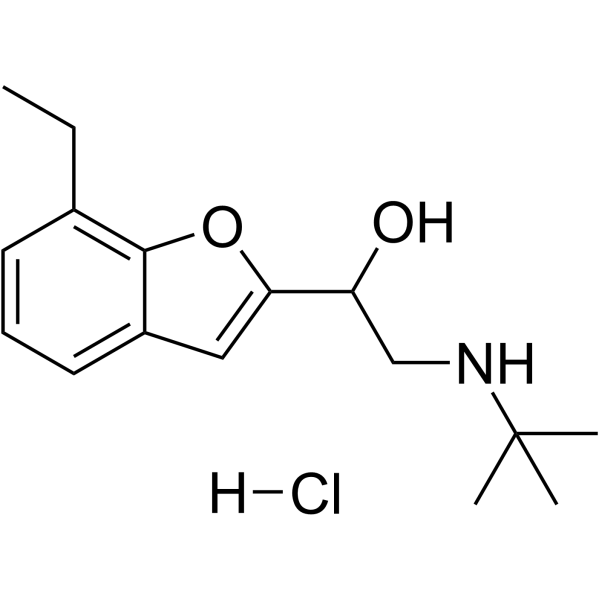
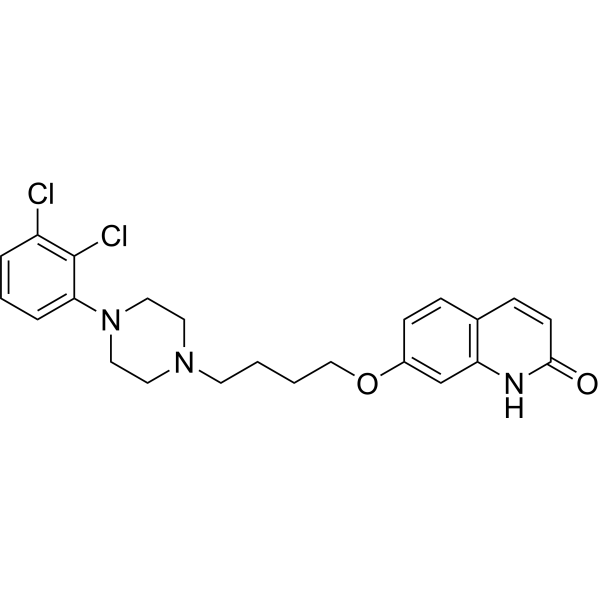
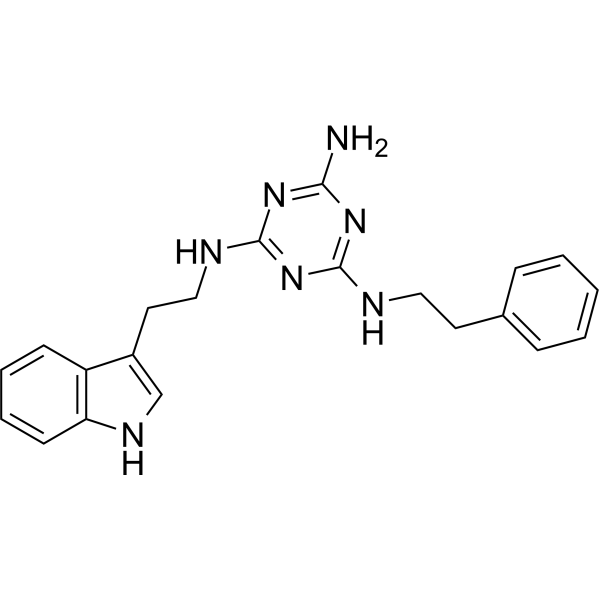
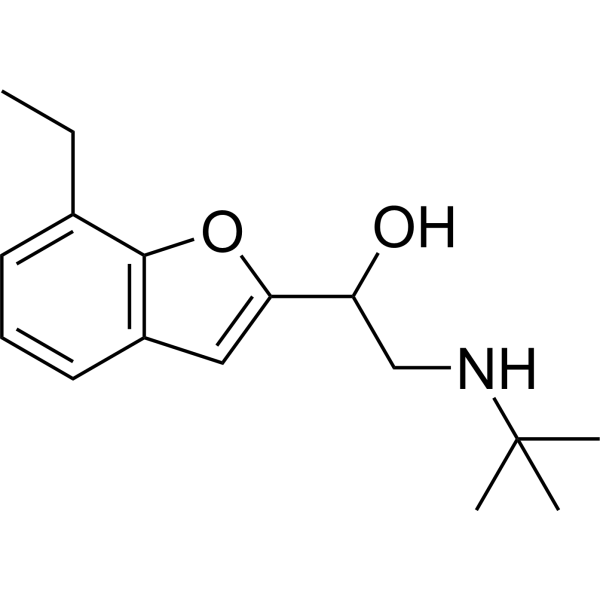
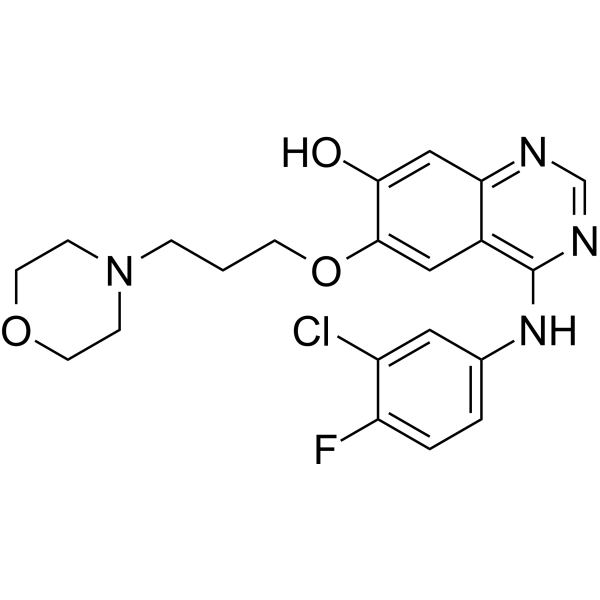
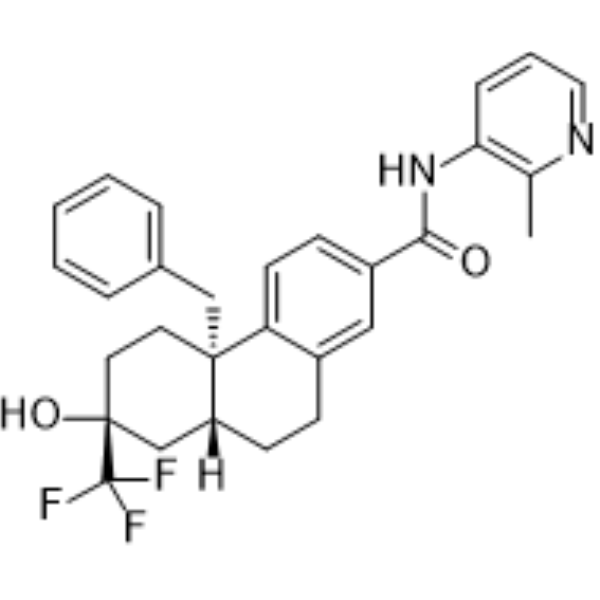
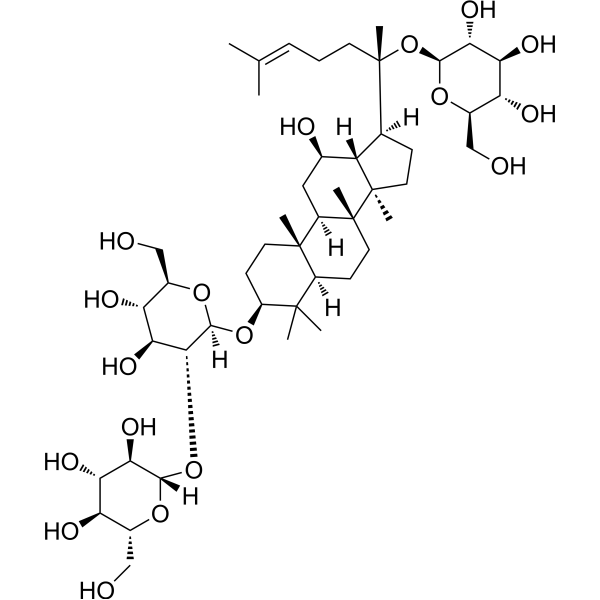
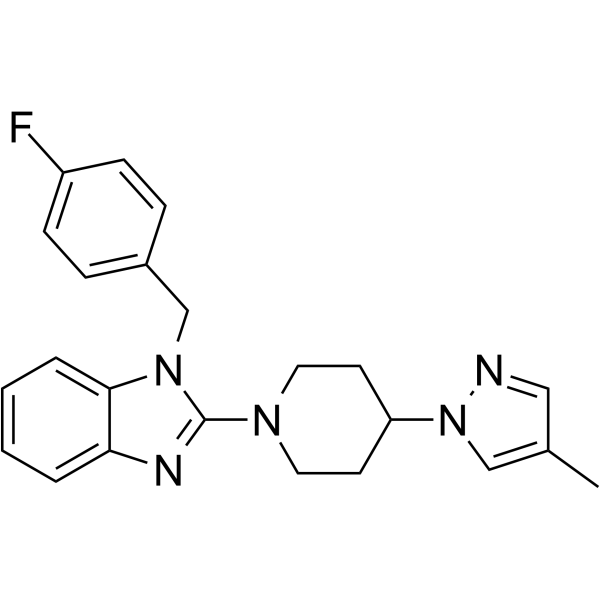
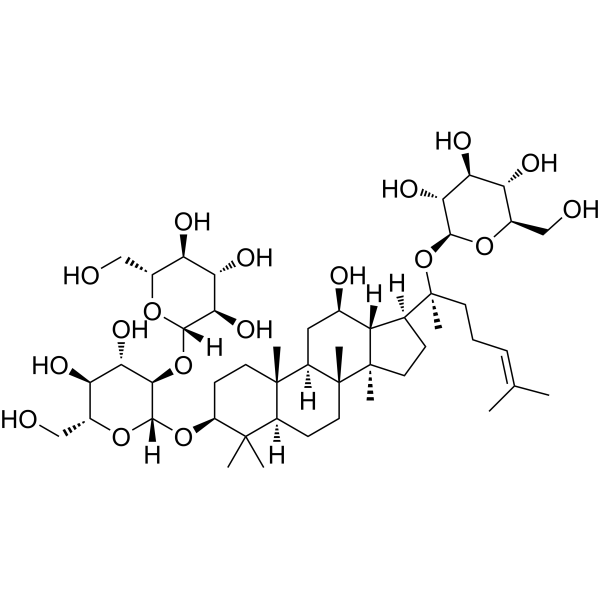
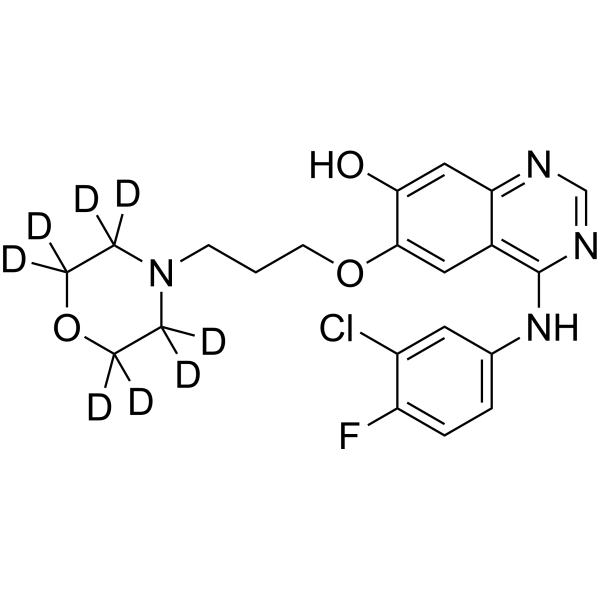
From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
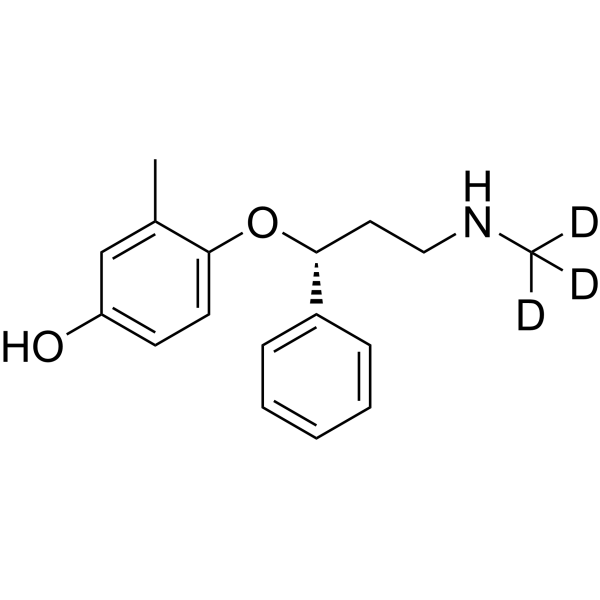
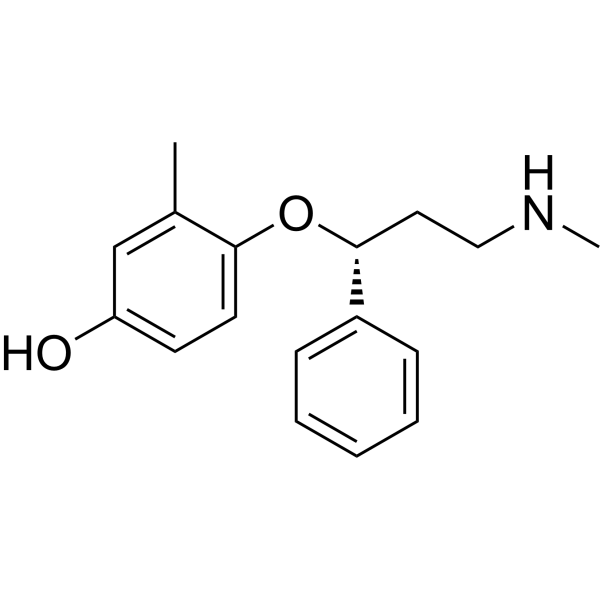
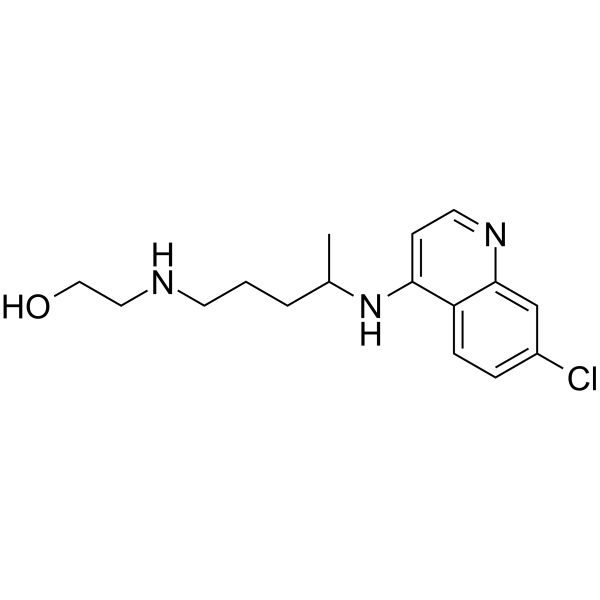
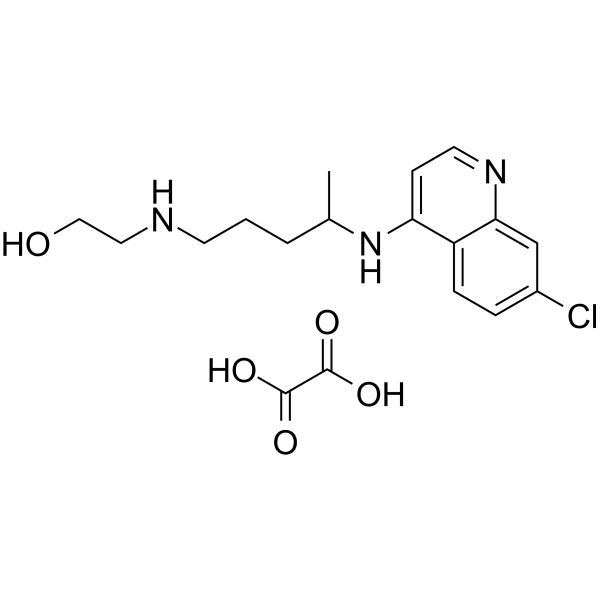
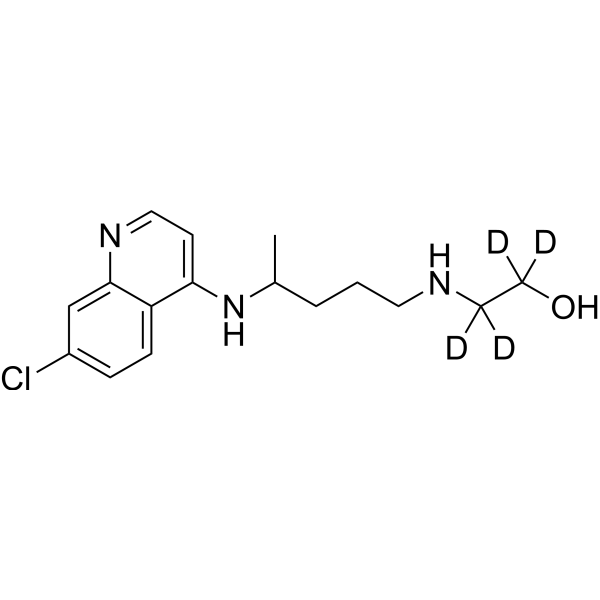
4-Hydroxyatomoxetine-d3 is a deuterium labeled 4-Hydroxyatomoxetine. 4-Hydroxyatomoxetine is an active metabolite of Atomoxetine (Tomoxetine). 4-Hydroxyatomoxetine is metabolized by the enzyme cytochrome P450 2D6 (CYP2D6). Atomoxetine is a potent and selective noradrenal in re-uptake inhibitor[1].
CYP2D6 Human Pre-designed siRNA Set A contains three designed siRNAs for CYP2D6 gene (Human), as well as a negative control, a positive control, and a FAM-labeled negative control.
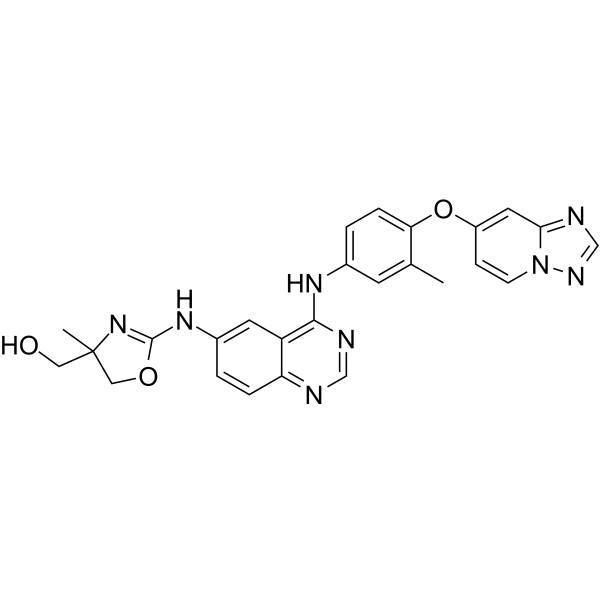
CXCR4 antagonist 3 (compound 12a) is a potent antagonist of CXCR4 with an IC50 of 11 nM. CXCR4 antagonist 3 is a congener of TIQ15. CXCR4 antagonist 3 demonstrates the best overall properties including CXCR4 antagonism, CYP2D6 inhibition, metabolic stability, and permeability. CXCR4 antagonist 3 has the potential for the research of human immunodeficiency virus .
CXCR4 antagonist 4 is a potent, orally active CXCR4 antagonist (IC50=24 nM) with diminished CYP2D6 activity, improved PAMPA permeability, potent inhibition of human immunodeficiency virus entry (IC50=7 nM) .
4-Hydroxyatomoxetine is an active metabolite of Atomoxetine. 4-Hydroxyatomoxetine is metabolized by the enzyme cytochrome P450 2D6 (CYP2D6). Atomoxetine hydrochloride is a potent and selective noradrenalin re-uptake inhibitor (Ki values are 5 nM, 77 nM and 1451 nM for inhibition of radioligand binding to human NET, SERT and DAT respectively) .
Norfluoxetine hydrochloride is an active N-demethylated metabolite of Fluoxetine. Fluoxetine is a selective serotonin (5-HT) reuptake inhibitor that is metabolized to Norfluoxetine hydrochloride by cytochrome P450 (CYP) 2D6, CYP2C19, and CYP3A4. Norfluoxetine hydrochloride inhibits 5-HT uptake and inhibits CaV3.3 T current (IC50 = 5 μM). Norfluoxetine hydrochloride has anticonvulsant activity .
LKY-047, a Decursin derivative, is a potent and selective reversible competitive cytochrome P45022J2 (CYP2J2) inhibitor with an IC50 of 1.7 μM. LKY-047 is inactive against other human P450s, such as CYPs 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A .
Chalepensin, a furanocoumarin, is a competitive CYP2A6 inhibitor. Chalepensin also inhibits human CYP1A1, CYP1A2, CYP2A13, CYP2C9, CYP2D6, CYP2E1, and CYP3A4 to different extents .
Peucedanol is a non-competitive inhibitor of CYP3A4 with a Ki value of 4.07 μM and a competitive inhibitor of CYP1A2 and CYP2D6 with Ki values of 3.39 μM and 6.77 μM, respectively .
CYP3A4 enzyme-IN-1 (compound 59) is a potent antibacterial agent, with a MIC of 1 μg/mL for MRSA. CYP3A4 enzyme-IN-1 exhibits low to moderate inhibitory effects on CYP1A2, CYP2E1, CYP2D6, and CYP3A4 enzymes .
Ginsenoside F1, an enzymatically modified derivative of Ginsenoside Rg1, demonstrates competitive inhibition of CYP3A4 activity and weaker inhibition of CYP2D6 activity.
HIV-IN-9 (Compound 2b) is a HIV inhibitor (IC50: 6.65 μg/mL), and has high binding affinity with HIV-RT. HIV-IN-9 also inhibits CYP3A4, CYP1A2, CYP2C1, and CYP2D6 .
Thermopsoside is a flavone derivative isolated from Aspalathus linearis. Thermopsoside exhibits inhibitory effects on CYP450 isozymes with IC50 values of 6.0 μM, 9.5 μM, 12.0 μM, 32.0 μM, for CYP3A4, CYP2C19, CYP2D6 and CYP2C9, respectively .
Rhodiosin is a double inhibitor of CYP2D6 and AChE, and can be isolated from Rhodiolis rhodiolis root. The IC50 for CYP2D6 is 0.761 μM, and the Ki is 0.769 μM. Rhodiosin has antioxidant and neuroprotective activity and can regulate HIF-1α signaling pathway to protect the central nervous system
Cletoquine (Desethylhydroxychloroquine) is a major active metabolite of Hydroxychloroquine. Cletoquine is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine has antimalarial effects and has the potential for autoimmune diseases treatment .
Dehydroaripiprazole (OPC-14857) is an active metabolite of Aripiprazole. Aripiprazole is an antipsychotic agent and is metabolized by CYP3A4 and CYP2D6 forming mainly Dehydroaripiprazole. Dehydroaripiprazole has with antipsychotic activity equivalent to Aripiprazole .
Dehydroaripiprazole (OPC-14857) hydrochloride is an active metabolite of Aripiprazole. Aripiprazole is an antipsychotic agent and is metabolized by CYP3A4 and CYP2D6 forming mainly Dehydroaripiprazole hydrochloride. Dehydroaripiprazole hydrochloride has with antipsychotic activity equivalent to Aripiprazole .
ONT-993 is an aliphatic hydroxylated metabolite. ONT-993 inhibits CYP2D6 (IC50=7.9 µM) and causes metabolism-dependent inactivation of CYP3A (KI=1.6 µM) .
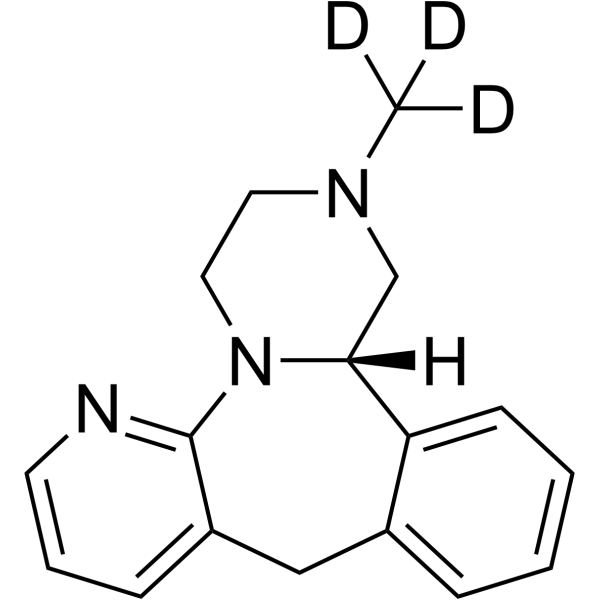
(S)-Mirtazapine-d3 is a deuterium labeled (S)-Mirtazapine. (S)-Mirtazapine is a S(+)-enantiomer of Mirtazapine with pronociceptive properties in an animal model of acute thermal nociception.(S)-Mirtazapine is a stereoselective 5-HT2 receptor antagonist. (S)-Mirtazapine is metabolized by CYP2D6 and CYP1A2[1].
Cletoquine oxalate (Desethylhydroxychloroquine oxalate) is a major active metabolite of Hydroxychloroquine. Cletoquine oxalate is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine oxalate is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine oxalate has antimalarial effects and has the potential for autoimmune diseases treatment .
Guanfu base A is an antiarrhythmic alkaloid isolated from Aconitum coreanum and is a potent noncompetitive CYP2D6 inhibitor, with a Ki of 1.20 μM in human liver microsomes (HLMs) and a Ki of 0.37 μM for the human recombinant form (rCYP2D6). Guanfu base A is also a potent competitive inhibitor of CYP2D in monkey (Ki of 0.38 μM) and dog (Ki of 2.4 μM) microsomes . Guanfu base A also inhibits HERG channel current .
Cletoquine-d4 is deuterium labeled Cletoquine. Cletoquine (Desethylhydroxychloroquine) is a major active metabolite of Hydroxychloroquine. Cletoquine is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine has antimalarial effects and has the potential for autoimmune diseases treatment[1][2].
(S)-Mirtazapine ((S)-Org3770) is a S(+)-enantiomer of Mirtazapine with pronociceptive properties in an animal model of acute thermal nociception. (S)-Mirtazapine is a stereoselective 5-HT2 receptor antagonist. (S)-Mirtazapine is metabolized by CYP2D6 and CYP1A2 .
Cletoquine-d4-1 is the deuterium labeled Cletoquine. Cletoquine (Desethylhydroxychloroquine) is a major active metabolite of Hydroxychloroquine. Cletoquine is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine has antimalarial effects and has the potential for autoimmune diseases treatment[1][2].
Dehydroaripiprazole-d8 is deuterium labeled Dehydroaripiprazole. Dehydroaripiprazole (OPC-14857) is an active metabolite of Aripiprazole. Aripiprazole is an antipsychotic agent and is metabolized by CYP3A4 and CYP2D6 forming mainly Dehydroaripiprazole. Dehydroaripiprazole has with antipsychotic activity equivalent to Aripiprazole[1][2][3].
2-Phenyl-2-(1-piperidinyl)propane is a selective and reversible human CYP2B6 inhibitor with an IC50 of 5.1 μM and a Ki of 5.6. 2-Phenyl-2-(1-piperidinyl)propane inhibits CYP2D6 (IC50=74 μM), CYP3A (IC50=200 μM) .
Bufuralol (Ro 3-4787) hydrochloride is a potent non-selective, orally active β-adrenoreceptor antagonist with partial agonist activity. Bufuralol hydrochloride is a CYP2D6 probe substrate .
Dehydroaripiprazole (Standard) is the analytical standard of Dehydroaripiprazole. This product is intended for research and analytical applications. Dehydroaripiprazole (OPC-14857) is an active metabolite of Aripiprazole. Aripiprazole is an antipsychotic agent and is metabolized by CYP3A4 and CYP2D6 forming mainly Dehydroaripiprazole. Dehydroaripiprazole has with antipsychotic activity equivalent to Aripiprazole .
5-HT7 receptor ligand 1 (Compound 5c) is a 5-HT7 receptor ligand with a Ki of 8 nM. 5-HT7 receptor ligand 1 is not hepatotoxic and exhibit moderate potential interaction with other agents metabolized by CYP3A4 or CYP2D6 .
Bufuralol (Ro 3-4787) is a potent non-selective, orally active β-adrenoreceptor antagonist with partial agonist activity. Bufuralol hydrochloride is a CYP2D6 probe substrate .
O-Desmethyl gefitinib is an active metabolite of Gefitinib in human plasma. The formation of O-desmethyl gefitinib is dependent on CYP2D6 activity. O-desmethyl gefitinib inhibits EGFR with an IC50 of 36 nM in subcellular assays .
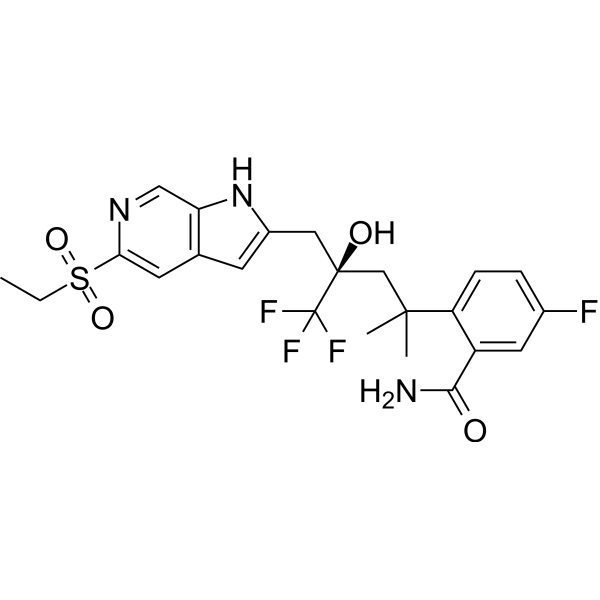
Dagrocorat (PF-00251802) is an orally active and selective high-affinity partial agonist of the glucocorticoid receptor. Dagrocorat is also a time-dependent reversible inhibitor of CYP3A (IC50=1.3 μM in human liver microsomes) and CYP2D6 (Ki=0.57 μM in human liver microsomes). Dagrocorat can be used for the research of rheumatoid arthritis .
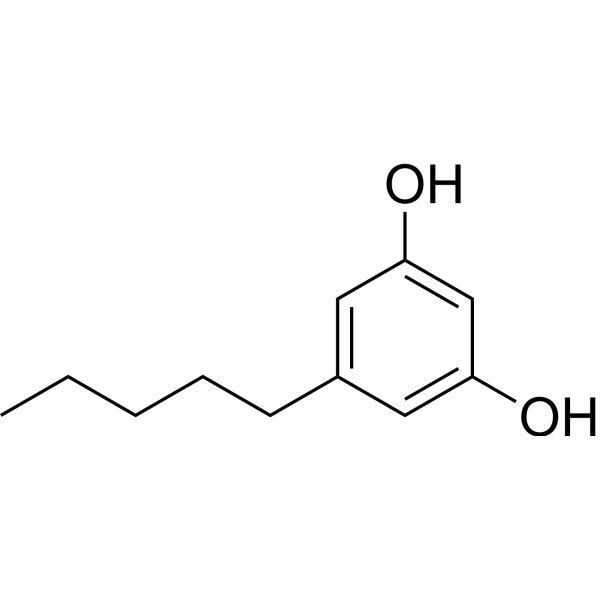
Olivetol is a naturally phenol found in lichens and produced by certain insects, acting as a competitive inhibitor of the cannabinoid receptors CB1 and CB2 . Olivetol also inhibits CYP2C19 and CYP2D6 activity, with IC50s of 15.3 μM, 7.21 μM and Kis of 2.71 μM, 2.87 μM, respectively .
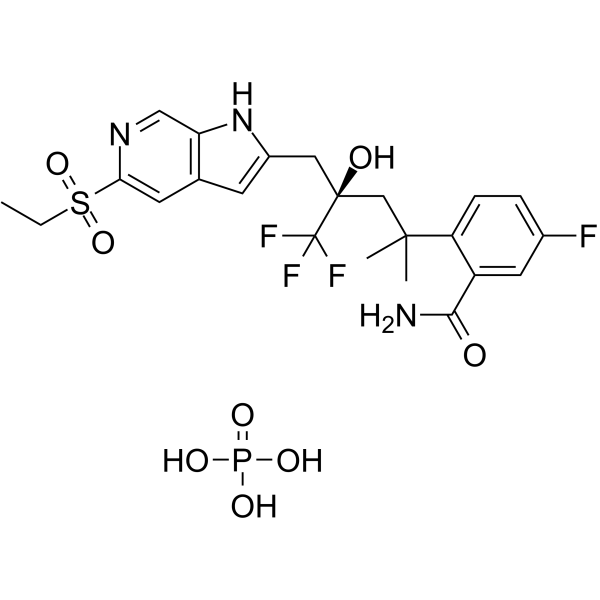
Dagrocorat (PF-00251802) hydrochloride is an orally active and selective high-affinity partial agonist of the glucocorticoid receptor. Dagrocorat hydrochloride is also a time-dependent reversible inhibitor of CYP3A (IC50=1.3 μM in human liver microsomes) and CYP2D6 (Ki=0.57 μM in human liver microsomes). Dagrocorat hydrochloride can be used for the research of rheumatoid arthritis .
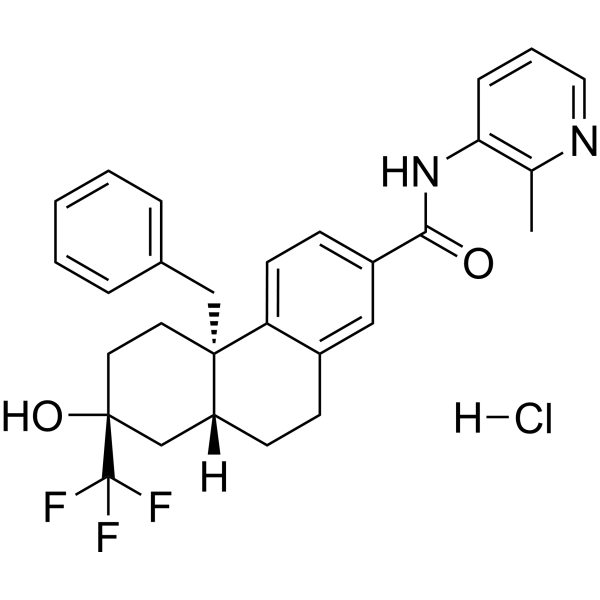
BI 653048 phosphate is a selective and orally active nonsteroidal glucocorticoid (GC) agonist with an IC50 value of 55 nM . BI 653048 phosphate inhibits CP1A2, CYP2D6, CYP2C9, CYP2C19 and CYP3A4 isoforms’ activity and reduces affinity for the hERG ion channel (IC50>30 μM) . BI 653048 phosphate is extracted from patent WO2005028501A1 (Compound 103), is also a HCV NS3 protease inhibitor that can reduce viral loads infected with the hepatitis C virus .
BI 653048 is a selective and orally active nonsteroidal glucocorticoid (GC) agonist with an IC50 value of 55 nM . BI 653048 inhibits CP1A2, CYP2D6, CYP2C9, CYP2C19 and CYP3A4 isoforms’ activity and reduces affinity for the hERG ion channel (IC50>30 μM) . BI 653048 is extracted from patent WO2005028501A1 (Compound 103), is also a HCV NS3 protease inhibitor that can reduce viral loads infected with the hepatitis C virus .
Ginsenoside Rd inhibits TNFα-induced NF-κB transcriptional activity with an IC50 of 12.05±0.82 μM in HepG2 cells. Ginsenoside Rd inhibits expression of COX-2 and iNOS mRNA. Ginsenoside Rd also inhibits Ca 2+ influx. Ginsenoside Rd inhibits CYP2D6, CYP1A2, CYP3A4, and CYP2C9, with IC50s of 58.0±4.5 μM, 78.4±5.3 μM, 81.7±2.6 μM, and 85.1±9.1 μM, respectively.
Antihistamine-1 is a H1-antihistamine (Ki=6.9 nM) with acceptable blood-brain barrier penetration and also an inhibitor of CYP2D6 and hERG channel with IC50s of 5.4 and 0.8 μM, respectively.
Ginsenoside Rd (Standard) is the analytical standard of Ginsenoside Rd. This product is intended for research and analytical applications. Ginsenoside Rd inhibits TNFα-induced NF-κB transcriptional activity with an IC50 of 12.05±0.82 μM in HepG2 cells. Ginsenoside Rd inhibits expression of COX-2 and iNOS mRNA. Ginsenoside Rd also inhibits Ca 2+ influx. Ginsenoside Rd inhibits CYP2D6, CYP1A2, CYP3A4, and CYP2C9, with IC50s of 58.0±4.5 μM, 78.4±5.3 μM, 81.7±2.6 μM, and 85.1±9.1 μM, respectively.
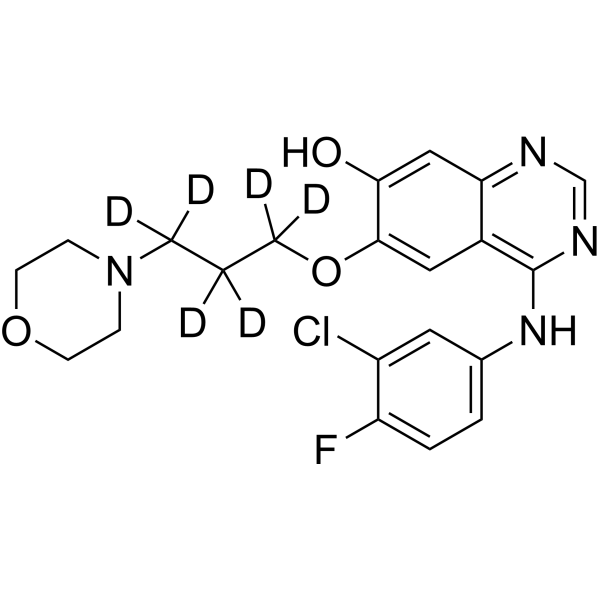
O-Desmethyl gefitinib-d8 is a deuterium labeled O-Desmethyl gefitinib. O-Desmethyl gefitinib is an active metabolite of Gefitinib in human plasma. The formation of O-desmethyl gefitinib is dependent on CYP2D6 activity. O-desmethyl gefitinib inhibits EGFR with an IC50 of 36 nM in subcellular assays[1][2].
O-Desmethyl gefitinib-d6 is the deuterium labeled O-Desmethyl gefitinib. O-Desmethyl gefitinib is an active metabolite of Gefitinib in human plasma. The formation of O-desmethyl gefitinib is dependent on CYP2D6 activity. O-desmethyl gefitinib inhibits EGFR with an IC50 of 36 nM in subcellular assays[1][2].
GSK-25 is a potent, selective and orally bioavailable ROCK1 inhibitor (IC50=7 nM). GSK-25 maintains good selectivity against a panel of 31 kinases (>100 fold), as well as RSK1 and p70S6K (RSK1: IC50=398 nM, p70S6K: IC50=1 μM). GSK-25 inhibits P450 profile (IC50s of 2.5, 5.2, 2.5 µM for CYP2C9, CYP2D6, CYP3A4, respectively) .
CXCR4 antagonist 4 is a potent, orally active CXCR4 antagonist (IC50=24 nM) with diminished CYP2D6 activity, improved PAMPA permeability, potent inhibition of human immunodeficiency virus entry (IC50=7 nM) .
Ginsenoside F1, an enzymatically modified derivative of Ginsenoside Rg1, demonstrates competitive inhibition of CYP3A4 activity and weaker inhibition of CYP2D6 activity.
Chalepensin, a furanocoumarin, is a competitive CYP2A6 inhibitor. Chalepensin also inhibits human CYP1A1, CYP1A2, CYP2A13, CYP2C9, CYP2D6, CYP2E1, and CYP3A4 to different extents .
Peucedanol is a non-competitive inhibitor of CYP3A4 with a Ki value of 4.07 μM and a competitive inhibitor of CYP1A2 and CYP2D6 with Ki values of 3.39 μM and 6.77 μM, respectively .
Thermopsoside is a flavone derivative isolated from Aspalathus linearis. Thermopsoside exhibits inhibitory effects on CYP450 isozymes with IC50 values of 6.0 μM, 9.5 μM, 12.0 μM, 32.0 μM, for CYP3A4, CYP2C19, CYP2D6 and CYP2C9, respectively .
Rhodiosin is a double inhibitor of CYP2D6 and AChE, and can be isolated from Rhodiolis rhodiolis root. The IC50 for CYP2D6 is 0.761 μM, and the Ki is 0.769 μM. Rhodiosin has antioxidant and neuroprotective activity and can regulate HIF-1α signaling pathway to protect the central nervous system
Guanfu base A is an antiarrhythmic alkaloid isolated from Aconitum coreanum and is a potent noncompetitive CYP2D6 inhibitor, with a Ki of 1.20 μM in human liver microsomes (HLMs) and a Ki of 0.37 μM for the human recombinant form (rCYP2D6). Guanfu base A is also a potent competitive inhibitor of CYP2D in monkey (Ki of 0.38 μM) and dog (Ki of 2.4 μM) microsomes . Guanfu base A also inhibits HERG channel current .
Olivetol is a naturally phenol found in lichens and produced by certain insects, acting as a competitive inhibitor of the cannabinoid receptors CB1 and CB2 . Olivetol also inhibits CYP2C19 and CYP2D6 activity, with IC50s of 15.3 μM, 7.21 μM and Kis of 2.71 μM, 2.87 μM, respectively .
Ginsenoside Rd inhibits TNFα-induced NF-κB transcriptional activity with an IC50 of 12.05±0.82 μM in HepG2 cells. Ginsenoside Rd inhibits expression of COX-2 and iNOS mRNA. Ginsenoside Rd also inhibits Ca 2+ influx. Ginsenoside Rd inhibits CYP2D6, CYP1A2, CYP3A4, and CYP2C9, with IC50s of 58.0±4.5 μM, 78.4±5.3 μM, 81.7±2.6 μM, and 85.1±9.1 μM, respectively.
Ginsenoside Rd (Standard) is the analytical standard of Ginsenoside Rd. This product is intended for research and analytical applications. Ginsenoside Rd inhibits TNFα-induced NF-κB transcriptional activity with an IC50 of 12.05±0.82 μM in HepG2 cells. Ginsenoside Rd inhibits expression of COX-2 and iNOS mRNA. Ginsenoside Rd also inhibits Ca 2+ influx. Ginsenoside Rd inhibits CYP2D6, CYP1A2, CYP3A4, and CYP2C9, with IC50s of 58.0±4.5 μM, 78.4±5.3 μM, 81.7±2.6 μM, and 85.1±9.1 μM, respectively.
The CYP2D6 protein, a vital cytochrome P450 monooxygenase, metabolizes diverse substrates, including fatty acids and steroids. It catalyzes epoxidation, hydroxylation, and oxidative conversion, impacting endocannabinoid signaling, cholesterol homeostasis, and drug metabolism, highlighting its multifunctional role in cellular processes. CYP2D6 Protein, Human is the recombinant human-derived CYP2D6 protein, expressed by E. coli , with tag free. The total length of CYP2D6 Protein, Human is 464 a.a., .
The CYP2D6 protein, a vital cytochrome P450 monooxygenase, metabolizes diverse substrates, including fatty acids and steroids. It catalyzes epoxidation, hydroxylation, and oxidative conversion, impacting endocannabinoid signaling, cholesterol homeostasis, and drug metabolism, highlighting its multifunctional role in cellular processes. CYP2D6 Protein, Human (His) is the recombinant human-derived CYP2D6 protein, expressed by E. coli , with N-6*His labeled tag. The total length of CYP2D6 Protein, Human (His) is 464 a.a., .
4-Hydroxyatomoxetine-d3 is a deuterium labeled 4-Hydroxyatomoxetine. 4-Hydroxyatomoxetine is an active metabolite of Atomoxetine (Tomoxetine). 4-Hydroxyatomoxetine is metabolized by the enzyme cytochrome P450 2D6 (CYP2D6). Atomoxetine is a potent and selective noradrenal in re-uptake inhibitor[1].
(S)-Mirtazapine-d3 is a deuterium labeled (S)-Mirtazapine. (S)-Mirtazapine is a S(+)-enantiomer of Mirtazapine with pronociceptive properties in an animal model of acute thermal nociception.(S)-Mirtazapine is a stereoselective 5-HT2 receptor antagonist. (S)-Mirtazapine is metabolized by CYP2D6 and CYP1A2[1].
Cletoquine-d4 is deuterium labeled Cletoquine. Cletoquine (Desethylhydroxychloroquine) is a major active metabolite of Hydroxychloroquine. Cletoquine is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine has antimalarial effects and has the potential for autoimmune diseases treatment[1][2].
Cletoquine-d4-1 is the deuterium labeled Cletoquine. Cletoquine (Desethylhydroxychloroquine) is a major active metabolite of Hydroxychloroquine. Cletoquine is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine has antimalarial effects and has the potential for autoimmune diseases treatment[1][2].
Dehydroaripiprazole-d8 is deuterium labeled Dehydroaripiprazole. Dehydroaripiprazole (OPC-14857) is an active metabolite of Aripiprazole. Aripiprazole is an antipsychotic agent and is metabolized by CYP3A4 and CYP2D6 forming mainly Dehydroaripiprazole. Dehydroaripiprazole has with antipsychotic activity equivalent to Aripiprazole[1][2][3].
O-Desmethyl gefitinib-d8 is a deuterium labeled O-Desmethyl gefitinib. O-Desmethyl gefitinib is an active metabolite of Gefitinib in human plasma. The formation of O-desmethyl gefitinib is dependent on CYP2D6 activity. O-desmethyl gefitinib inhibits EGFR with an IC50 of 36 nM in subcellular assays[1][2].
O-Desmethyl gefitinib-d6 is the deuterium labeled O-Desmethyl gefitinib. O-Desmethyl gefitinib is an active metabolite of Gefitinib in human plasma. The formation of O-desmethyl gefitinib is dependent on CYP2D6 activity. O-desmethyl gefitinib inhibits EGFR with an IC50 of 36 nM in subcellular assays[1][2].